New Human Physiology | Paulev-Zubieta 2nd Edition
Chapter 30: Other Hormones and Disorders
| HOME | PREFACE | TABLE OF CONTENTS | SYMBOLS | SECTION INFO | CONTRIBUTORS | LINKS | CONTACT US |
Highlights
Study_ObjectivesPrinciplesDefinitionsEssentials
PathophysiologyEquationsSelf-AssessmentAnswers
Further Reading
|
Chapter 30
|
 |
|
|
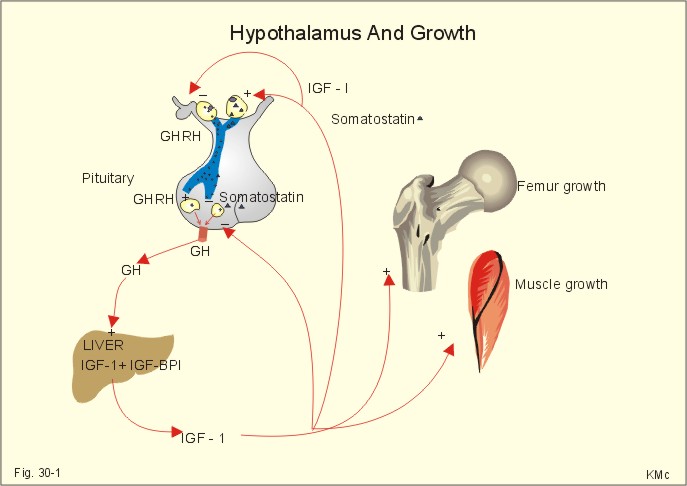
· To define or describe concepts such as catecholamines, nanism, pituitary dwarfism, pseudo-hypo-para-thyroidism, somatomedins, somatostatin, and somatotropin. · To describe the regulation of extracellular and intracellular Ca2+, the bone structure, remodelling and the function of osteoblasts, osteoclasts and osteocytes. To describe the biosynthesis of calcitonin, parathyroid hormone, vitamin D, mineralocorticoids, glucocorticoids, adrenaline and noradrenaline. To describe osteoporosis, osteomalacia, and rachitis. To describe the Ca2+ and phosphate balance in healthy persons. · To explain the effects of calcitonin, parathyroid hormone, vitamin D, mineralocorticoids, glucocorticoids, androgens and oestrogens from the adrenal cortex, and adrenaline/noradrenaline. To explain phenomena such as phaeochromocytoma, shock, adrenogenital syndrome, virilization and pubertas praecox. · To use the above concepts in problem solving and case histories. · The sympatho-adrenergic system give rise to the fright, flight or fight reactions that are all acute stress situations. · The adrenal cortex is concerned with the carbohydrate metabolism and with the electrolyte balance. · The parathyroid hormone and vitamin D maintain normal levels of calcium and phosphate in the body. · Calcium concentration (total) in plasma: 2.2-2.7 mM. Half of the total calcium is ionized. · Catecholamines are substances consisting of catechol (an aromatic structure with two hydroxyl groups) linked to an amine. The important catecholamines in humans are adrenaline, noradrenaline and dopamine. · Cushing’s disease is hypercorticism (increased glucocorticoid production) caused by a pituitary basophilic adenoma. · Cushing’s syndrome refers to the consequences of increased plasma glucocorticoid concentration from any source. · Growth hormone releasing hormone (GHRH) is a peptide produced by GHRH cells in the hypothalamus. GHRH stimulates the release of growth hormone (Somatotropin) from the adenohypophysis. · Hypercalcaemia is an increase in the plasma concentration of ionized calcium. The condition is characterised by neurological signs (hyporeflexia, lethargy or coma). When Ca2+ is deposited in the inner medulla of the kidney, ADH resistance develops with polyuria and polydipsia. · Hypocalcaemia is a decrease in the plasma concentration of ionized Ca2+ (cramps, tetany, twitching and tingling of the fingers). Hypocalcaemia is caused by renal failure or by thyroidectomy and parathyroidectomy. · Hyperparathyroidism. Primary hyperparathyroidism is caused by parathyroid adenomas or by hyperplasia. Secondary hyperparathyroidism is compensatory hypertrophia due to hypocalcaemia. · Hypoparathyroidism. The idiopathic form is a rare autoimmune destruction of the parathyroid (with hypocalcaemia). · Infantile nanism is a term used to indicate that the somatic age of a child is below its chronological age. · Nanism or dwarf growth expresses that the height of an adult is below a certain limit. In the Anglo-Saxon countries the limit is 1.40 m for females and 1.50 m for males. · Pituitary dwarfism refers to low pituitary secretion of GH or insensitive target organ receptors (African pygmies). · Pseudo-hypo-para-thyroidism is used for conditions, where target-organs (bone, kidneys, and gut) are resistant to PTH. The plasma- [Ca2+ ] is low, plasma-[phosphate] is high, and basic phosphatase activity is high. · Somatomedins are small peptides (5 kDa) produced in the liver, promote bone growth and protein synthesis. Somatomedin C is also called insulin-like growth-factor -1 (IGF-1). The hepatic production of somatomedins is stimulated by growth hormone. Somatomedins stimulate the hypothalamic secretion of somatostatin. · Somatotropin or growth hormone (GH) from the anterior pituitary stimulates body growth in humans. GHRH stimulates and somatostatin inhibits the release of GH. Growth hormone stimulates the hepatic production of Somatomedin C (IGF-1). · Somatostatin or growth hormone inhibiting hormone (GHIH) is a cyclic peptide, which is produced in the pancreatic islets and in the CNS, where it is co-transmitter with noradrenaline. GHIH inhibits the release of growth hormone from the adenohypophysis and is therefore also called somatotropin releasing inhibiting factor, SRIF. This paragraph deals with 1. Growth hormone, 2. Parathyroid hormone. Calcium and phosphate homeostasis, 3. Adrenal corticoids, 4. Adrenal catecholamines, and 5. Vitamin D and bone minerals. Growth hormone is the main stimulator of body growth in humans. Growth hormone is produced in the pituitary, stimulated by hypothalamic growth hormone releasing hormone (GHRH) and inhibited by hypothalamic somatostatin (Fig. 30-1). Pituitary growth hormone stimulates the hepatic production of transcription factors such as Somatomedin C (IGF-1) and its specific IGF-binding protein 1 (IGF-BP1). Actually, it is somatomedin C that stimulates body growth - both bone and muscle growth.
Fig. 30-1: Pituitary GH and hepatic IGF-1 secretion in the hypothalamic-pituitary feedback system. Growth hormone releasing hormone (GHRH) is produced by GHRH cells in the hypothalamus and reaches the adenohypophysis via the portal system. GHRH stimulates the release of growth hormone (GH) from the adenohypophysis (Fig. 30-1). Growth hormone and insulin are important anabolic hormones in humans, although growth hormone has many insulin-antagonistic effects. Growth hormone is released as hunger spikes during the day and during sleep. Growth hormone (GH) produced in the placenta differs from the pituitary GH by a few Amino acid residues. Placental GH suppresses release of maternal, pituitary GH during Pregnancy. Placental GH stimulates maternal metabolism and foetal cell proliferation and hypertrophia. Foetal thyroid hormones stimulate brain development, and foetal insulin stimulates foetal growth, cellular glucose uptake and glucose utilisation. Paracrine and autocrine growth Factors are also important for foetal growth: Insulin-like growth factor-II (IGF-II), nerve Growth factors (NGF), epidermal growth factor (EGF), and platelet derived growth factor (PDGF). The pituitary GH has an endocrine effect on the production of the growth factor Somatomedin C (IGF-1) and its specific binding protein in the liver. Hepatic IGF-1 circulates in plasma bound to IGF-binding protein 1 (IGF-BP1). The binding proteins are important, not only as a vehicle in plasma, but also for the final binding of the hormone to its cell membrane receptor. One GH binds to two cell receptor molecules, the intercellular portion of which activate thyrosine kinasis. The kinasis phosphorylate the transcription factors that modulate gene expression. Hepatic IGF-I stimulates bone formation, and GH stimulates the precondrocytes directly. Locally released IGF-I stimulates the condrocytes in healthy states, but it also has a regenerative function in damaged tissues. Pituitary GH also stimulates the production of receptors for other growth factors. GH serves to commit a precursor cell to a specific pathway, and IGF-I enhances its growth and replication. Somatomedins inhibit the secretion of GH from the somatotropic cells of the adenohypophysis. Somatomedins also stimulate the hypothalamic secretion of somatostatin (GHIH or somatotropin releasing inhibiting factor, SRIF). Tissue specific growth factors - just like those important for foetal growth - are produced in damaged tissues, where they are important for regeneration. Nervous tissues produce NGF, epidermal tissues produce EGF, thrombocytes produce PDGF, and fibroblasts produce both fibroblast growth factor (FGF) and transforming growth factors (TGF-a, and TGF-b). Hepatocyte proliferation after liver damage is induced by both hepatocyte growth factor (HGF) and by hepatocyte stimulating substance (HSS). Growth factors are used in regenerative therapy.
Fig. 30-2: 68-1: Growth curves in boys - a similar pattern is found in girls. The relative large growth of the testes at puberty shows the importance of sex hormones for early puberty growth (Fig. 30-2). In general, the growth spurt in early puberty is caused by increased production of sex steroids (stimulated by luteinizing hormone and by follicle stimulating hormone, FSH), because the sex steroids stimulate hypothalamus to release more growth hormone. Growth hormone and insulin are probably the most important anabolic hormones in the body - more effective than oestradiol and testosterone; however, GH has many insulin-antagonistic effects. Human GH stimulates protein synthesis, mitosis in cells, chondrogenesis, ossification, and phosphate balance (increased renal phosphate reabsorption), while increasing glycolysis (ie, anaerobic breakdown of glycogen). GH stimulates hepatic glucose production (glycogenolysis) but not its gluconeogenesis. GH enhances RNA synthesis, accelerates glucose uptake and antagonise the lipolytic effect of adrenaline. - GH is produced in spikes during the day (hunger spikes) and during deep sleep (EEG stage III and IV), and the half-life of GH is 20 min. Children with pan-hypopituitarism become infantile dwarfs and later turn into juvenile dwarfs, as they do not develop sexually. Children with hyperpituitarism become giantesses or giants. The clinical diagnosis is gigantismus. Hypothyroid children (cretins) also become dwarfs. The thyroid hormones are necessary or permissive for normal growth and development. Cretins are mentally retarded, hypothyroid dwarfs. Sex steroids in high concentrations close the epiphyseal lines. If this occurs early in life, the child also becomes an infantile dwarf, but such an individual is often sexually active. The clinical diagnosis is precocious puberty or pubertas praecox. Without proper insulin treatment, children with diabetes (mellitus) become dwarfs (diabetic, infantile nanismus), because of the intracellular hypoglycaemia. Poorly regulated diabetics also develop osteopenia (bone decalcification). Genetic factors are essential for optimal growth and development, as shown by the strong correlation between the height of the parents and the final height of the child. Tall people are high before the puberty growth spurt, which is more or less the same for tall and shorter people. - Persons with only one sex chromosome (X, 0) show retarded growth from birth, whereas persons with an extra sex chromosome (XXY; XYY) become tall. Optimal growth depends upon optimal nutrition (essential amino acids, vitamins, minerals, and fatty acids) and optimal Neuroendocrine, metabolic control. Optimal growth also depends upon an optimal health. Most disease states and all immuno-defence threatening treatments retard growth or imply weight loss. Such disorders cause catabolic hormones to dominate and induce anorexia. 2. Parathyroid hormone (PTH). Calcium and phosphate homeostasis. We overlooked the existence of the parathyroid glands until the consequence of their surgical removal was realised. Without the parathyroid, a person develops tetanic cramps due to a fall in the concentration of ionised calcium ([Ca2+]) in the blood plasma (explained in Chapter 17). - Already in 1909, MacCallum treated this condition successfully with calcium salts. The chief cells (C cells) of the parathyroid glands produce pre-proparathyroid hormone, which is cleaved in their endoplasmic reticulum to proparathyroid hormone. This in turn is cleaved in the Golgi apparatus to parathyroid hormone (PTH). PTH is a single chain peptide with a molecular weight of 9500 Da. PTH is water-soluble and binds to membrane receptors on the surface of the target cells. Thus the PTH action is dependent on second messengers. Both intracellular Ca2+ and cyclic adenosine monophosphate (cAMP) are used. Humans carry a single PTH gene on chromosome 11. The chief cells also produce a parathyroid secretory protein of unknown function. In man, the four parathyroid glands are located just behind the thyroid gland. Ectopic tissue sometimes develops in the mediastinum or in the neck. PTH binds to membrane receptors on all target cells. The major effects of PTH are on three target organs: 1. Bone: PTH accelerates the removal of Ca2+ and phosphate from bones (osteolysis by surface osteocytes, resorption of bone by osteoclasts). PTH stimulates the osteolysis by surface osteocytes causing the release of Ca2+ for rapid equilibrium with the ECV. After 12 hours, the delayed effect of PTH, which stimulates the osteoclasts to reabsorb mineralised bone, sets in. 2.Kidney: PTH reduces the reabsorption of Ca2+ and phosphate from the proximal tubules, and increases the reabsorption of Ca2+ from the distal tubules, frequently resulting in an increased net loss of Ca2+ in the urine. PTH binds to the basolateral membrane of the tubule cell, and stimulates cAMP, which in turn diffuses through the cell to the luminal membrane. Here cAMP activates a Ca2+ -reabsorption port. The glomerular filtration of Ca2+ is easy to calculate, since approximately half the total plasma concentration is free and filterable (2.5/2 mM). Since 0.94 parts of plasma is water, the [Ca2+] in the ultrafiltrate is (1.25/0.94) 1.33 mM. A person with an average glomerular filtration rate of 0.125 l per min will produce a 24-hour ultrafiltrate of 180 litre. Thus, a total Ca2+ flux of (1.33 × 180 =) 239 mmol or almost 10 000 mg daily, will pass the glomerular ultrafilter. Fortunately, almost all Ca2+ is reabsorbed in the kidney tubules (about 67% is reabsorbed in the proximal, and the reabsorption of the balance in the distal tubules is regulated by PTH). We only excrete 100 to 200 mg daily (or 2.5 to 5 mmol daily) in the urine and 50 mg or 1.1 mmol through the skin. This is a daily maximum of 250 mg (or 6 mmol) Ca2+ excretion from the body. Another drastic action of PTH is on the proximal tubules, where PTH inhibits phosphate reabsorption so efficiently that its excretion in the urine increases within 5 minutes. 3.Gut: The PTH action on the gut is indirect. PTH stimulates the renal production of biologically active vitamin-D (1,25-dihydroxy-vitamin D = calcitriol), which stimulates the active absorption of Ca2+ and phosphate across the gut mucosa (Fig. 30-3), and potentiates the action of PTH on bone resorption.
Fig. 30-3: The normal calcium transfer (all numbers are in mg per day or in [mmol per day]). The balance is 250 mg per day. The end result of these three actions is an increase in plasma [Ca2+ ] and a decrease in plasma [phosphate]. Simultaneously, the bone resorption activity is illustrated by a high basic phosphatase concentration. Cystic areas, corresponding to periostal reabsorption, are visible on bone radiographs (osteitis fibrosa cystica). Hypercalcaemia predisposes to kidney stones and to metastatic calcification in synovial membranes and meninges, in the lungs, kidneys, pancreas and elsewhere. Certain tumours produce PTH-related protein with sequence homology to PTH. Perhaps this substance is related to the hypercalcaemia of malignant processes (see ectopic tumours). The daily need of phosphate is 18 mmol just as the daily need of Ca2+ .The human body contains 25 mol of phosphorous (31 Da) as phosphate. Out of the 25 mol, 20 mol is located in bones and about 4 mol in muscle and other soft tissues (Fig. 30-4). A daily food intake of 46 mmol of phosphate (equal to the Ca2+ intake) with a secretion of 3 mmol and an intestinal absorption of 39 mmol, leaves (10 + 36) mmol daily for faecal and renal excretion in order to be in balance. The normal plasma concentration of phosphate is 1 mM. This implies a daily phosphate filtration rate (net-flux) of 180 mmol (1 mM*180 l of plasma) - at a glomerular filtration rate of 0.125 l per min. Phosphate is a threshold substance with a tubular reabsorption capacity (Tmax) of 0.1 mmol per min; this means that 80% of the filtered load is reabsorbed. The maximal daily filtration- reabsorption- and excretion flux is calculated in Fig. 30-4. Most of the reabsorption of phosphate takes place in the proximal tubules, where PTH inhibits phosphate reabsorption. Renal control maintains the phosphate concentration in blood plasma. The Tmax for phosphate is up regulated by high phosphate intake and down regulated by low phosphate intake in the food. The daily exchange of phosphate from bone is 8 mmol and from soft tissue 16 mmol (Fig. 30-4). – Phosphate depletion may leads to muscle weakness (cardiac and skeletal muscles) and pathological bone formation.
Fig. 30-4: The daily phosphate balance. The binding of PTH activates adenylcyclase and this raises the [cAMP], which interacts with protein kinase A. The enzyme then catalyses the phosphorylation of effector proteins. PTH is secreted in response to hypocalcaemia, in particular, a low [Ca2+] (or low [Mg2+]). The major effects of PTH are to increase plasma [Ca2+] and decrease plasma [phosphate] through its effect on three target organs: bone, kidney and gut. – Magnesium Mg2+ is a cofactor in enzyme reactions and important for the neuromuscular transmission. The adrenal cortex of the adult human has three layers: The outer zona glomerulosa consists of small cells and is narrow, the middle zona fasciculata is wide, and the inner zona reticularis contains a reticulum of interconnected cells. The adrenal cortex is of mesodermal origin. Steroids The human adrenal cortex produces three types of steroids: Glucocorticoids, mineralocorticoids, and a minimal amount of sex steroids (androgens and oestrogens). All steroids are lipid-soluble and easily cross the lipid membrane. All steroids represent chemical modifications of four-ring structures (A-D) forming the cyclopentano-perhydro-phenantren. Steroids bind to specific cytosol-receptor proteins that are then translocated to the cell nucleus. Here they reversibly bind to DNA. The synthesis of glucocorticoids (cortisol and corticosterone) occurs in the zona fasciculata with a small contribution from zona reticularis. The mineralocorticoid, aldosterone, is produced in no other region of the cortex than zona glomerulosa. The synthesis of sex hormones (androgens and oestrogens) occurs mainly in zona reticularis. The precursor for these hormones is cholesterol absorbed from the blood HDL and LDL fractions by the cortex cells. Most of the synthetic reactions involve mixed oxygenases (belonging to the cytochrome P-450 enzymes) localized in the endoplasmatic reticulum and in the mitochondria. During ACTH stimulation the size and number of cells in the zona fasciculata and zona reticularis increase, mainly because the cortisol and the sex hormone production increase. The mitochondria, central ribosomes, vesicular cristae and endoplasmic reticulum grow in these cells. ACTH activates all steps in corticosteoid hormone synthesis. A microsomal desmolase removes C20-21 from the precursors, pregnenolone and progesterone (C21 steroids). The residues are dehydroepi-androsterone and androstenedione (C19). These androgens are weak and are converted to a more potent form, testosterone, in peripheral tissues. In the zona reticularis, testosterone is converted to oestradiol (C18), due to removal of a CH3 group by aromatase. The gene for the human androgen receptors is found on the X chromosome. The receptor protein has a molecular weight of 98 kDa and is found both in the cytosol and the nucleus. CRH & ACTH The hypothalamic Corticotropin Releasing Hormone (CRH) stimulates the secretion of ACTH. Stress stimulates not only the sympatho-adrenergic system with catecholamine release, but also the CRH/ACTH release with increased secretion of neuroregulatory peptides and cortisol. This is important, since small amounts of cortisol have permissive effects on catecholamines, while inhibiting TSH. Stress also releases growth hormone (GH that stimulates glycogenolysis/glycolysis) and prolactin, both from the acidophilic cells in the adenohypophysis. Prolactin released by stress is possibly mediated by hypothalamic histaminergic neurons. ACTH binds to the cells of zona fasciculata and activates adenylcyclase, which results in a rise in the cAMP level. The major effect of ACTH - through increased cAMP level - is to stimulate the conversion of cholesterol to pregnenolone by desmolase. This is the rate-limiting step in the production of cortical steroids! Plasma levels of cortisol (hydrocortisone), adrenal androgens and their precursors rise within three minutes of intravenous ACTH injection. Cortisol inhibits the release of CRH and ACTH by negative feedback. Glucocorticoids Kendall isolated cortisone, and Hench & Reichstein directed the first administration of cortisone and ACTH to patients with rheumatoid arthritis. The result was a dramatic improvement. They shared the Nobel Prize in 1950. The serious side effects were recognized later. The gene for the human glucocorticoid receptor is present in chromosome 5. The receptor protein (94 kDa) is found in both the cytosol and the nucleus.The effects of a glucocorticoid such as cortisol are physiologic or pharmacological dependent upon the dose. Cortisol is the main endogenous glucocorticoid synthesized in the adrenal cortex. Cortisol is essential for life and acts permissively to facilitate the mobilisation of fuels. Cortisol defends the body against hypoglycaemia evoked by insulin. Cortisol stimulates hepatic glucose production (gluconeogenesis). Cortisol augments the glucagon stimulation of glycogenolysis. Cortisol inhibits the glucose uptake in target cells (GLUT 4 in muscle cells, heart cells and adipocytes). Cortisol is diabetogenic. Cortisol is lipolytic and acts permissively on adrenaline, GH and other lipolytic substances to mobilize triglycerides. Cortisol also suppresses CRH release and an excess of cortisol may lead to truncal obesity. Cortisol induces leptin synthesis in fat cells. This synthesis limits the appetite by negative feedback. Cortisol maintains the contractility of striated and cardiac muscle (the Na+- K+-pump, b-adrenergic receptors etc). Excess cortisol increases muscle metabolism and reduces muscle mass and strength. Cortisol decreases the ratio of insulin-sensitive slow oxidative type I muscle fibres to the fast glycolytis type II-B muscle fibres. Cortisol reduces the differentiation to active osteoblasts and the synthesis of collagen in bone matrix and connective tissue. Cortisol antagonises the action of 1,25-dihydroxy-cholecalciferol and thus the absorption of Ca2+ from the gut – cortisol excess leads to osteoporosis. Cortisol is permissive to the vasoconstriction of catecholamines and angiotensin II. Cortisol increases renal bloodflow and GFR. Cortisol is important for the development of CNS, skin, gut, bone marrow and lungs. Glucocorticoids stimulate erythropoiesis. Cortisol inhibits processes involved in inflammation, infections, tissue damage, and immune system reactions. Glucocorticoids inhibit most responses mediated by leucotrienes, NO, PAF, prostaglandins, and thromboxanes. Therapeutic doses of glucocorticoids are used to treat more diseases than any other group of drugs, because the hormones act anti-inflammatory and anti-allergic. The negative effects are delayed healing of wounds and increased gluconeogenesis with destruction of tissue proteins. In plasma, most of the cortisol binds to transcortin, to corticosteroid binding globulin (CBG), or to albumin and only 5% is free. The protein binding means that cortisol has a long plasma-T½ of more than 70 min. Oestrogens stimulate the production of cortisol binding proteins. Patients with liver and kidney diseases do not produce enough. – Transcortin-cortisol complexes activate adenylyl cyclase, and the complex liberates free colesterol at sites of inflammation. A low concentration of hormone specific globulin implies a low concentration of the bound hormone fraction, but the free hormone concentration can still be the same. Cortisol is converted into tetrahydrocortisol, cortisone or to 17-ketosteroids. Cortisone is a biologic analog to cortisol. Exogenous cortisone is an important source of cortisol in many tissues due to the presence of the enzyme, 11beta-hydroxy-dehydrogenase. Conjugation with glucuronic acid or sulphate forms water-soluble products. Only minimal amounts of the daily cortisol secretion is excreted in the urine. The fraction excreted in the bile is released to the enterohepatic circuit.Half of the daily cortisol secretion is excreted in the urine as 17-hydroxycorticoids. The plasma cortisol concentration is increased by anxiety, burns, fever, exercise, hypoglycaemia, pain, severe physical or psychiatric disease, stress and surgery.The plasma cortisol concentration decreases promptly following administration of synthetic glucocorticoids (dexamethasone), which suppress ACTH secretion by negative feedback. Mineralocorticoids Aldosterone is the major mineralocorticoid with corticosterone contributing only little. Aldosterone maintains ECV by conserving body Na+. When body Na+ is depleted, the fall in ECV and plasma volume reduces renal bloodflow and pressure. A reduction in circulating fluid volume recorded in the kidneys releases aldosterone. Aldosterone promotes the reabsorption of Na+ and increases the secretion of K+ and H+ in the distal tubules and the cortical collecting ducts. A rise in plasma-[K+] from normal or a fall in plasma-[Na+] also releases aldosterone by direct action on zona glomerulosa. The renin-angiotensin-aldosterone cascade (Chapter 24) controls the adrenal aldosterone secretion, not the ACTH. Only a small fraction of the aldosterone binds to proteins. This makes its half-life short (20 min). Aldosterone is reduced to tetra-hydro-aldosterone in the liver and is conjugated with glucuronic acid. Aldosterone like all other steroids is lipophilic, so that it passes easily through the cell membrane. The human mineralocorticoid receptor is found primarily in the cytoplasm, and its gene is present on chromosome 4. This receptor has been cloned revealing a molecular weight of 107 kDa. When aldosterone is bound to the receptor, the receptor reveals a DNA-binding site. The receptor-aldosterone complex translocates from the cytosol to the nucleus, where it binds to chromatin by means of the DNA-binding site. This process activates the transcription of the specific gene producing mRNA, which is then translated into specific proteins. Following complete transmission, the receptor-aldosterone complex dissociates from chromatin and each other. The receptor returns to the cytosol with a hidden DNA-binding site. By this process aldosterone promotes the synthesis of new proteins, that may stimulate the Na+-K+-pump or facilitate Na+ entry into the tubular cell through integral Na+ -channel proteins in the luminal membrane. Increased Na+-reabsorption from the tubules due to aldosterone causes a simultaneous reabsorption of water and thus an increased ECV, with increased arterial blood pressure. The pressure rise leads to increased GFR. The rapid filtration flow overrides the high reabsorptive effect of aldosterone, which down-regulates the size of ECV. – Beta-adrenergic blockers lower the arterial pressure in part by reducing the Na+ -retention caused by the renin cascade. Inhibitors of angiotensin converting enzyme or angiotensin II receptor blockers also lower the arterial blood pressure. Antagonists of aldosterone at the renal distal tubule cells directly prevent Na+ -reabsorption and reduce hypertension. The ACTH effect is only a moderate stimulation in acute situations. The action of angiotensin II on aldosterone secretion is much more important. The adenohypophysis probably produces an aldosterone-stimulating factor. Dopamine (Prolactin inhibiting hormone, PIH) is an aldosterone inhibitor. Adrenal sex corticoids are mainly weak adrenal androgens produced largely in zona reticularis. By conversion to testosterone and dihydrotestosterone in peripheral tissues, the adrenal androgens play a physiological role. Also a small oestrogen production is present in healthy persons. In females, adrenal androgens produce testosterone for normal pubic and axillary hair development and erythrogenesis. The medulla is a modified sympathetic ganglion derived from neuroectodermal cells. The development of the sympathoadrenergic nervous system is stimulated by neural growth factors. The adrenal medulla is the source of the circulating catecholamine, adrenaline. The medulla also secretes small amounts of nor-adrenaline, normally a neurotransmitter. The sympathetic system consists of short preganglionic fibres, which have tropic centers in the lateral horn of the spinal cord from the first thoracic segment to the third lumbar segment. The myelinated nerve fibres form ramus communicans albus and pass to the paravertebral or lateral sympathetic ganglion chain. Here most of the fibres end on the postganglionic cell bodies. Some preganglionic fibres pass the sympathetic chain and reach collateral ganglia such as the solar plexus, and the ganglion mesentericus superior and inferior. In these lateral and collateral relay stations each preganglionic fibre is in contact with many cell bodies. These cells have long, postganglionic, unmyelinated fibres serving the different organs. The preganglionic fibres to the adrenal medulla pass all the way to the chromaffin cells in the adrenal medulla. These chromaffin cells (high affinity for chromium cell stains) are embryological analogues to postganglionic fibres and the synapse is cholinergic. Synthesis of catecholamines Catecholamines are substances consisting of catechol (an aromatic structure with two hydroxyl groups) linked to an amine. The catecholamines are synthesises within the chromaffin cell. The dietary amino acid tyrosine is absorbed as tyrosine from the gut (Fig. 30-5). Tyrosine is also produced in the liver from dietary phenylalanine. The L-tyrosine is hydroxylated by tyrosine hydroxylase in the cytosol of the chromaffin cell, and the product is L-dihydroxy-phenyl-alanine (Dopa, Fig. 30-5). Tyrosine hydroxylase is the rate-limiting step in the catecholamine production. Dopa is decarboxylated to dopamine, catalysed by Dopa-decarboxylase. Dopamine is produced in the cytosol, and then taken up by the chromaffin granules, which contain the enzyme, dopamine b-hydroxylase. The enzyme catalyses the formation of nor-adrenaline from dopamine. In most granules noradrenaline diffuses back into the cytosol, where it is N-methylated by the enzyme, phenyl-ethanolamine N-methyl-transferase to form adrenaline. Adrenaline is then stored in the granules as the most important adrenomedullary hormone. The storage process requires ATP, and catecholamines are stored with ATP in the granules. - In the newborn the primary end product in the medulla is nor-adrenaline, but with advancing age there is a dramatic rise in the adrenaline content. This conversion depends upon cortisol. The three important catecholamines in humans are dopamine, epinephrine or adrenaline (Ad), and norepinephrine or noradrenaline (NA in Fig. 30-5). Dopa is converted to melanin in the melanocytes of the skin, stimulated by melanocytic stimulating hormone (MSH in Fig. 30-5).
Fig. 30-5: Synthesis of catecholamines within the chromaffin cell. Tyrosine hydroxylase and dopamine b-hydroxylase is activated by sympathetic stimulation. Catecholamines are formed in adrenergic, noradrenergic and dopaminergic neurons. The neurons of substantia nigra produce dopamine, which by axonal transport reaches the striatum (corpus striatum in Fig. 30-5). Here, dopamine is stored in the granules of the terminals - ready for liberation in the synapses. In the sympathetic ganglion cells, dopamine is just an intermediary step, which is oxidised (dopamine b-hydroxylase) to form noradrenaline. Euler identified noradrenaline as the chemical transmitter from adrenergic nerves, and Axelrod demonstrated the reuptake of the transmitter after its release from nerve terminals. In 1970 they shared the Nobel Prize with Katz, who explained the role of calcium in synaptic transmission. Actions of catecholamines Catecholamines act on adrenergic membrane receptors designated a1-, a2-, b1- b2- and b3-receptors. The a-receptors are located on effector cells that are highly sensitive to noradrenaline, less so to adrenaline, but much less so to isoprenaline (a synthetic catecholamine). The b-receptors are located on effector cells that are most sensitive to isoprenaline, but less so to adrenaline (and noradrenaline). The a1-receptor acts via calcium and protein kinase C. The a2-receptor acts through G-protein inhibition of membrane adenylyl cyclase. The b-receptors are transmembrane glycoproteins, and they use cAMP as second messenger. Catecholamines prevent hypoglycaemia and restore glucose delivery to the CNS. Catecholamines increase glucose production by gluconeogenesis in the liver, and stimulate glycogenolysis in liver and muscle cells. In the absence of glucagon, adrenaline is important for recovery from hypoglycaemia. Adrenaline inhibits the insulin-mediated glucose uptake by muscle and fat cells. Catecholamines stimulate glucagon secretion and inhibit insulin secretion. Adrenaline activates lipase in fat cells and thus increases FFA release, b-oxidation of FFA in muscle and liver and ketogenesis. Adrenaline increases basal metabolic rate, nonshivering thermogenesis, and diet-induced thermogenesis during cold exposure. Catecholamines stimulate proton transfer into the mitochondria and uncouple ATP synthesis from oxygen utilisation. Catecholamines increase the heart rate, contractile force and the cardiac output by stimulation of the adrenergic b1-receptors in the myocardium. Noradrenergic nerve fibres innervate vessels all over the body, and this system usually has some tonic, vasoconstrictor activity. The a1-receptors are located on the surface of vascular smooth muscles. Adrenaline constricts the arterioles in the cutaneous, renal and splancnic beds. Catecholamines dilatate the bronchial airways by stimulation of their adrenergic b2-receptors. They increase both tidal volume and respiratory frequency. The result is an increased ventilation with an increased cardiac output. Catecholamines relax the smooth muscles of the digestive tract (b2-receptors), but contract the sphincters just like the sympathetic nerve system. Finally, adrenaline stimulates the ascending reticular system (ie, the reticular activating system or RAS) in the brain stem, keeping us alert and causing arousal reactions with desynchronisation of the EEG. Stress responses Stress comprises severe emotional and physical burdens (fear, pain, hypoxia, hypothermia, hypoglycaemia, hypotension etc). The adrenal medulla functions in concert with the adrenal cortex during stress and during immune system disorders. The two systems provide useful local cytokines and avoid systemic damages from accumulated cytokines. Stress activates the CRH-ACTH-corticoid axis and adrenergic neurons in the hypothalamus with connections to the sympathetic nervous system. ACTH is released whereby plasma cortisol is elevated; the adrenergic stimulus elevates plasma adrenaline. These hormones increase glucose production. Noradrenaline and CRH induce a state of arousal and aggressiveness. CRH inhibits sexual activity and feeding behaviour. The sympatho-adrenergic system gives rise to the fright, flight or fight-reactions, which are all acute stress situations. During stress adrenaline induces hyperglycaemia and ketoacidosis (a diabetogenic hormone). Prolonged stress liberates ACTH via hypothalamic signals. ACTH stimulates the glucocorticoid secretion through cAMP. Small amounts of glucocorticoids are permissive for the actions of catecholamines. Acute stress activates the splancnic nerves and liberates large amounts of adrenaline from the medulla. Diabetics, who are developing acute hypoglycaemia, secrete large amounts of catecholamines. In the endangered individual catecholamines dilatate bronchioles, inhibit gastrointestinal motility, and dilatate pupils to improve distant vision. Exercise Catecholamines support the sympathetic system in modifying the circulation during exercise. During exercise catecholamines promote the use of muscle glycogen, the gluconeogenesis from lactate, and the mobilisation of FFA as alternative fuel. During exercise the blood is directed to the working muscles from the other parts. The resistance vessels of the striated muscles in hunting predators (and perhaps in humans) are also innervated by another system. This is the cholinergic, sympathetic vasodilator system. It is capable of a rapid and appropriate bloodflow response during hunting. The fall in the a-adrenergic tone in the muscular arterioles is probably the most important exercise response in humans. Metabolism of catecholamines Plasma catecholamines are rapidly removed from the blood and have half-lives in plasma of 20-60 s. This is the combined result of rapid uptake by tissues and inactivation in the liver and vessel walls. Enzymes inactivate catecholamines by methylation or by oxidation. Catechol-O-methyl-transferase (COMT) on the surface of the target cells catalyses methylation to metanephrine, normetanephrine. Monoamine oxidase (MAO) in the mitochondria catalyses oxidative removal of the amino group with formation of vanillylmandelic acid (VMA) and methoxy-hydroxy-phenyl-glycol (MOPG). The final products in the urine areVMA and MOPG. The normal excretion of VMA and MOPG in the daily urine averages 4 and 2 mg. 5. Vitamin D and bone minerals Bones are compared to iron mixed with concrete. The elasticity is due to the collagen (iron) network, and the stiffness (concrete) is due to calcium salts (Ca - hydroxyapatite complex). - Bone tissue consists of two compartments, bone cells (metabolically active) and the bone matrix (metabolically inert extracellular compartment). The organic part of the bone matrix consists of collagen fibres and ground substance, and the inorganic part consists of calcium-phosphate-hydroxyapatite. The atomic weight of calcium is 40, and we consume 1000 mg (25 mmol) per day. However, we absorb only 400 mg in total. The net-absorption is only 250 mg, since we secrete 150 mg daily to the intestines. Thus, 750 mg (19 mmol) must be excreted in the faeces every day (Fig 30-3). The net-absorption is saturable (an active process), since it depends on available Ca2+ -binding protein in the brush border and in the cytosol of the enterocyte. The synthesis of this protein in the mucosal cells, and thus intestinal Ca2+ -absorption, is induced by active vitamin D and by the parathyroid hormone. Steroid hormones like vitamin D, exerts their major effects after binding to specific nuclear receptors and then stimulating the synthesis of mRNA that codes for cytosolic Ca2+ -binding protein. The basolateral membrane contains two transporters of Ca2+: A Na+/Ca2+ exchanger, which is more effective at high intracellular [Ca2+], and a Ca2+-ATPase, which is the major mechanism at low levels of intracellular [Ca2+]. The duodenum-jejunum can concentrate Ca2+ against a tenfold concentration gradient. The amount of phosphate absorbed and its concentration in plasma is determined by the amount available in the diet, but the active transport is somewhat dependent on vitamin D. High plasma [Ca2+] and [phosphate] promote bone formation in children. The children increase the precipitation of Ca-hydroxy-apatite in their bone matrix. Intracellular phosphate is an essential component of nucleic acids, high-energy molecules, cofactors, regulatory phospho-proteins, and glycolytic intermediates. The [total calcium] in the blood plasma of healthy persons is normally 2.5 mM or 100 mg per l. Half the calcium binds to plasma proteins and 5% binds to a citrate-phosphate-bicarbonate complex. A significant portion (45-50%) is free ionised calcium ([Ca2+]); this part has critical roles in neuromuscular function and coagulation. If the plasma [Ca2+] falls to half its normal level, the body develops increased neural excitability with tetanic cramps (see below). On the other hand, an increase in [Ca2+] leads to calcification of soft tissue with heart, kidney and intestinal diseases. The [Ca2+] is the one variable regulated by parathyroid hormone (PTH) - see Fig. 30-3. The normal adult contains 27 500 mmol or 1100 g of calcium and 600 g of phosphate. The major part exists in the bones (1080 g fixed as hydroxyapatite and only 4 g or 100 mmol as exchangeable bone calcium). We exchange this 4 g of calcium five (5) times a day. The flux between the bones and the ECV (16 l) is approximately 500 mg or 13 mmol daily. This fast exchange regulates [Ca2+] in plasma. But we also have a small amount of daily bone formation and resorption (bone remodulation). In our soft tissue cells 99% of all calcium is complexed with phosphate in the mitochondria (10-5 mM), or bound to membranes and the endoplasmic reticulum. The cytosol contains an extraordinarily low [Ca2+]: 10-7 mol per l. The intracellular [Ca2+] is of extreme physiologic importance as regulator of enzymatic reactions and secretion, as well as a secondary messenger for peptide hormones. In general Ca2+ is important to almost all biological systems: action and membrane potentials, blood clotting, cell division, cellular secretion, contraction, cytoskeletal rearrangements, and motility. Both calcium and phosphate ions are in equilibrium with calcium-proteinate: Protein + Ca2+ => 2 H+ + Calcium-proteinate A hyperventilating patient eliminates carbonic acid, liberates H+ and reduces [Ca2+] in plasma, because more Ca2+ binds to protein. Low [Ca2+] in plasma leads to increased excitability of neuromuscular tissue and to tetany (tetanic muscle cramps). The treatment is simply to reinhale the exhaled air (rebreathing) resulting in carbonic acid accumulation (Chapter 17). – As seen from the reaction above, alkalosis reduces and acidosis increases [Ca2+] in plasma.
Fig. 30-6: Vitamin D and the Ca2+ balance. Vitamin D3 (cholecalciferol) from the skin, and vitamin D2 (ergocalciferol) from plant diet are concentrated in the liver. Both are produced in the skin by ultraviolet irradiation (Fig. 30-6). In the liver microsomal oxidase simply hydroxylates the molecules to the weakly active 25-hydroxy-cholecalciferol (25-OH-D). Lack of vitamin D leads to insufficient bone formation, because the osteoid matrix does not calcify. This disease is called rickets (rachitis) in children and osteomalacia in adults. This substance is transferred with the blood to the kidneys, where it is further hydroxylated at the C-1 position to the most potent form calcitriol (1,25-dihydroxy- cholecalciferol Fig. 30-6). This is what makes vitamin D a potent steroid hormone. Vitamin D is stored in muscle and adipose tissue and circulates in plasma bound to vitamin D-binding protein. Vitamin D2 is derived from vegetable ergosterol and its chemical name is ergocalciferol. Vitamin D3 or cholecalciferol is produced in the skin by ultraviolet irradiation. The two molecules have equal biological capacity in humans. Vitamin D must be hydroxylated to the weakly active 25-hydroxy-cholecalciferol in the liver. This substance is transferred with the blood to the liver, where it is further hydroxylated at the C-1 position to the most potent form 1, 25-dihydroxy-cholecalciferol. This is exactly what is necessary to make vitamin D a potent steroid hormone. Vitamin D circulates in plasma bound to vitamin D-binding protein and is stored in liver, muscle and adipose tissue. Vitamin D deficiency causes rickets in children and osteomalacia in adults. The deficiency is caused by insufficient diet, inadequate absorption as in fat malabsorption, insufficient synthesis in the skin, or abnormal conversion to its potent form in the liver and kidneys. Rickets (rachitis) is extremely rare in the rich part of the world. The epiphyseal plate of the growing skeleton is inefficiently mineralised and considerably thickened. Rachitic children have a flat skull with prominent frontal bones. The sternum protrudes as pigeon breast or pectus carinatum. The costal cartilage is enlarged forming the rachitic rosary. The limbs are shortened and deformed. Osteomalacia or soft bones are inadequate mineralisation of the organic bone matrix. Patients are asymptomatic or they suffer from diffuse pains. Bone X-rays are osteopenic. High plasma concentrations of parathyroid hormone, phosphate, and oestrogens stimulate the renal hydroxylation to active vitamin D. The active vitamin D-metabolite, the 1,25-dihydroxy-cholecalciferol, stimulates the transport of Ca2+ across the cell and mitochondrial membranes, and has the following two effects: 1. Enhanced effect on gut absorption of Ca2+, so that plasma-[Ca2+] increases, 2. Enhanced effect of PTH on bones. In vitamin D deficiency the low plasma-[Ca2+] stimulates the parathyroid glands, which leads to secondary hyper-parathyroidism. Ca2+ loss continues for months following space flight. Stimulation of bone deposition requires the stress of muscular activity as when a person is working against a gravity field. Appropriate exercise during space missions reduces the total Ca2+ loss. Puberty growth is essentially stimulated by the increased production of sex hormones, because the sex steroids stimulate hypothalamus to secrete more GH. Testosterone and oestradiol are generally important anabolic hormones. GH and insulin are also important anabolic hormones in humans, although GH has many insulin-antagonistic effects. GH is released as hunger spikes during the day and during sleep. Primary hormones for bone remodelling are GH, PTH, calcitonin and 1,25-D3. Secondary hormones for bone remodelling are glucocorticoids, sex hormones, thyroid hormones, prostaglandins (PGE2), insulin and IGF-I to III (somatomedins). Bone remodelling is a balance between bone formation by osteoblasts and bone resorption by osteoclasts and mononuclear cells. This balance involves the following hormones besides GH: 1. 1,25-Dihydro-cholecalciferol (1,25-D3) is a D-vitamin, but also a steroid hormone. Kidneys produce 1,25-D3 when stimulated by PTH. The steroid hormone increases the calcium absorption from the gut and mobilises Ca2+ from the bones. This increases the [Ca2+] and [phosphate] in plasma. The 1,25-D3 also increases the renal reabsorption of Ca2+ indirectly. 2. PTH also increases the plasma [Ca2+]. PTH mobilises Ca2+ from the bones and increases the reabsorption of Ca2+ in the distal, renal tubule cells while inhibiting phosphate reabsorption in the proximal tubules of the kidneys. 3. Calcitonin from the thyroid inhibits bone resorption by blocking the PTH receptors on the osteoclasts. Thus plasma [Ca2+] and [phosphate] fall. Cyclic treatment with calcitonin or combined treatment with 1,25-D3 and PTH improve bone formation in osteoporosis (ie, bone atrophia involving both minerals and matrix). Postmenopausal osteoporosis is treated successfully with calcitonin. Calcitonin is important in bone remodelling and in treatment of osteoporosis. A parallel to the bone formation by osteoblasts is the dentin formation by odontoblasts in our teeth. Dentists stimulate dentin formation with Ca(OH)2, when treating caries. This paragraph deals with: 1. Acromegaly and gigantism, 2. Nanism, 3. Hyperparathyroidism, 4. Hypoparathyroidism, 5. Hyperaldosteronism (Conn´s disease), 6. Cushings disease and syndrome, 7. Congenital adrenal hyperplasia, 8. Primary hypoadrenalism, 9. Secondary hypoadrenalism, 10. Phaeochromocytoma. Pituitary acidophilic adenomas that secrete excess growth hormone (GH) causes gigantism in children and acromegaly in adults. Rare cases are caused by GHRH excess release from the hypothalamus. Since pituitary acidophilic adenomas often contain somatotropic as well as mammotropic cells, the combined adenomas secrete an excess of both GH and prolactin (causing galactorrhoea in males). GH excess before the long bone epiphyses have closed, results in gigantism. The arms and legs are long, and the patients are tall. Giants may suffer from deficient sexual development, if the gonadotropic pituitary function also suffers. The hypophyseal tumour is demonstrated by CT scans or by magnetic resonance imaging. GH stimulates both soft-tissue and skeletal growth. In adults only bones in the cranium, the face, and the hands and feet respond the GH excess. Acromegaly often develops around the age of 30 years in both females and males.
Fig. 30-7: Clinical features of acromegaly. The calvarium, the maxilla and the mandible grow (Fig. 30-7). The teeth are separated by spaces and may fall out. The hands and feet are large and spade-shaped. This is why the adult disorder is called acromegaly (from Greek: extremity-great). Visual field defects are present when the enlarged hypophysis damages the crossed fibres in chiasma opticum, resulting in bilateral, heteronymous hemianopsia (Fig. 5-6). The GH concentration in plasma is only increased during stress and as occasional peak values, but IGF-1 is increased, and impaired glucose tolerance is diagnostic for a secondary, diabetic condition. There is often polyuria and glucosuria (Fig. 30-7). Therapy in cases with growing and pressure-damaging tumours is trans-sphenoidal or trans-frontal surgery with post-operative radiotherapy. Medical treatment with octreotide, an analogue to somatostatin (GHIH), is beneficial in some cases. Nanism or dwarf growth expresses that the height of an adult is below a certain limit. In the Anglo-Saxon countries the limit is 1.40 m for females and 1.50 m for males. Infantile nanism is a term used to indicate that the somatic age of a child is below its chronological age. Low pituitary secretion of GH or insensitive target organ receptor (African pygmies’) result in pituitary dwarfism. Many cases of dwarfism are idiopathic (of unknown origin). An achondroplastic dwarf has short limbs and a large head with a flat nose, because of abnormal growth of cartilage and bone. The abnormal gene is autosomal and dominant. Primary parathyroid hyperfunction is almost inevitably due to parathyroid adenomas or hyperplasia that secrete an excess of parathyroid hormone, PTH. Ectopic tumours have been found in the mediastinum and elsewhere. Excessive secretion of PTH leads to: bone resorption, high [Ca2+] in plasma, high Ca2+ -excretion in the kidneys with renal stone formation, bone lesions, and metastatic calcification (Fig. 30-8). The action of PTH on bone results in osteitis fibrosa cystica, there is hypercalcaemia with low plasma phosphate, metabolic acidosis and raised PTH levels in plasma. Secondary hyperparathyroidism is an adequate physiological response to hypocalcaemia by any cause such as renal failure or malabsorption. Hypercalcaemia has many other causes: hypervitaminosis D, excessive Ca2+ -intake, leukaemia, myelomata, sarcoidosis and cancer with bone metastases or malignant tumours producing PTH.
Fig. 30-8: Clinical manifestations of hyperparathyroidism. Hypoparathyroidism is characterised by hypocalcaemia, but most cases of hypocalcaemia are caused by renal failure with phosphate retention, vitamin D deficiency or resistance to vitamin D, resistance to PTH, pregnancy and lactation, or calcitonin administration. Most cases of hypoparathyroidism are iatrogenous (ie, caused by doctors during thyroidectomy or parathyroidectomy). After seemingly successful surgery there is a dramatic fall in plasma [Ca2+] and a rise in [phosphate], leading to hypocalcaemia cramps. The patient is in tetany with paresthesia, convulsions, and laryngeal stridor. Without therapy death ensues. Primary, idiopathic hypoparathyroidism is an extremely rare autoimmune disease often found in combination with other autoimmune disorders. Pseudo-hypo-para-thyroidism is used for conditions, where target-organs (bone, kidneys, and gut) are resistant to PTH. The plasma- [Ca2+ ] is low, plasma-[phosphate] is high, and basic phosphatase activity is high. A PTH injection does not increase renal phosphate excretion, because the kidneys are resistant. Hypoparathyroidism is treated with Ca2+ -infusion and the continuation is a lifelong treatment with PTH and vitamin D. 5. Hyperaldosteronism (Conn´s disease) Patients with adenoma or hyperplasia of the zona glomerulosa secrete large amounts of aldosterone - they suffer from primary hyperaldosteronism. Here, aldosterone works directly on the highly Na+ -loaded distal tubules, so the patient becomes severely K+-depleted. The renal retention of salt and water leads to hypertension. Loss of K+ and H+ leads to hypokalaemia with muscle weakness and cardiac arrhythmias and to metabolic alkalosis with hypocalcaemia and tetany. Plasma aldosterone is elevated although the patient has a high intake of NaCl. Removal of the neoplasm cures Conn´s disease. Secondary hyperaldosteronism. A patient with serious congestive heart failure may develop secondary hyperaldosteronism and become K+-depleted. At rest the GFR, the renal bloodflow, and the cardiac output are close to normal. The cardiac patient has an abnormally high Na+ retention and the accompanying high water retention that increases the ECV. This leads to oedema and increased venous return to the heart with a small rise in cardiac output. During exercise the rise in cardiac output is insufficient. The insufficient rise in bloodflow and blood pressure elicits (via the baroreceptors) a marked vasoconstriction of the renal circulation. Thus the RBF must decline. Other vascular beds also constrict markedly during exercise. The key factor to the abnormal Na+ retention is the reduction in RBF. The major Na+ reabsorption normally takes place in the proximal tubules. Therefore the distal tubular fluid contains a small load of Na+, allowing only a small K+ secretion here. This is in contrast to the patient with Conn's disease. 6. Cushings disease and syndrome Cushing’s disease is a pituitary disorder with increased secretion of pituitary ACTH. Cushing’s syndrome is used as a common term for all clinical cases of abnormally high glucocorticoid concentration, [cortisol], in the blood plasma. All clinical cases are divided into two groups: ACTH-dependent Cushing and non-ACTH-dependent Cushing. 1. ACTH-dependent Cushing is caused either by pituitary disorder (Cushing’s disease, see above) or by an ectopic ACTH-producing tumour. This group of cases is characterized by a high ACTH concentration in the plasma. 2. Non-ACTH-dependent Cushing. Most clinical cases are caused by glucocorticoid administration for long periods, but also adrenal tumours (adenomas and carcinomas) produce excess glucocorticoid. The ACTH secretion is suppressed. Clinical manifestations The high [cortisol ] in the blood has two major effects on the body: 1. Salt and water-retention with renal loss of K+ results in a plethoric tomato face or moon face (Fig. 30-9). The fluid-retention eventually leads to cardiac hypertrophy due to prolonged hypertension. There is often peripheral oedema, if not eliminated by polyuria due to the glucocorticoid-induced diabetes. This type of diabetes is typically resistant even to large doses of insulin. 2. Catabolism causes muscle wasting, fat accumulation, osteoporosis with kyphosis, buffalo hump, and fractures. The skin is thin with ulcers and red stirrer, and there is poor wound healing. There is impaired fibrocyte formation and capillary resistance.
Fig. 30-9: Manifestations of Cushing’s syndrome. In Cushings disease there is a pituitary tumour with enlarged sella turcica or pituitary hyperplasia. The pituitary corticotrophins produce excessive amounts of ACTH, which leads to bilateral adrenal cortical hyperplasia. In other cases of Cushing’s syndrome the primary cause is an adrenal tumour or adrenal hyperplasia. There is a hypersecretion in cortical zona reticularis and fasciculata. The patient may suffer from muscular and bone atrophy due to the catabolic effect of the cortisol surplus. The bone atrophy leads to osteoporosis, vertebral fractures and necrosis of the hips. There is a delayed healing of lesions and wounds due to the slow fibrocyte formation. The skin is thin with purple striae and fragile capillaries. Sodium and water accumulate in the body, whereas potassium is lost. The nitrogen balance is negative. The Cushing patient is often depressed and suffers from insomnia. The arms and legs are thin and weak due to the protein catabolic effect, but fat collects as truncal obesity and in the back and neck regions (bulls neck and buffalo hump). The fluid accumulation leads to a special look called moon face or tomato face. The high blood [cortisol] also increases the blood [glucose]. A diabetic condition, which is resistant to even large doses of insulin, develops. The patient suffers from hypertension and rapidly develops arteriosclerosis. Female patients complain of amenorrhoea and poor libido. The primary therapy is to reduce plasma [cortisol] to approximately 350 nM also as a preparation for surgery. In cases of Cushing’s disease the tumour is typically removed trans-sphenoidally, and adrenal adenomas are resected. The best assessment of increased ACTH production is by measuring the 24-hour urinary cortisol excretion. Suppression of ACTH and cortisol is performed with a standard dose of synthetic Glucocorticoid (dexamethasone), which is potent enough to suppress the ACTH production, without influencing the plasma [cortisol]. The principle of the suppression test is to challenge the hypothalamic-pituitary feedback system. If the system is intact a fall in blood [cortisol] is expected, and if not (hypothalamic-pituitary Cushing) the high [cortisol] is maintained. In healthy persons such a standard dose will suppress ACTH secretion, and lead to a reduced blood [cortisol]. Such a normal observation is also typical for patients with a primary adrenocortical hypertrophy. Dexamethazone does not suppress the high cortisol level of patients with a damaged feedback system producing excess ACTH from a pituitary tumour. Cortisol excess has been treated with ketoconazole, which inhibits several steps in the corticosteroid synthesis. Differential diagnosis: Alcoholic patients with so-called Pseudo-Cushing look exactly like Cushing’s syndrome, but the glucocorticoid concentration in the plasma is within normal limits. The cause is unknown. Cases with plasma [cortisol] in the upper end of the normal scale can be explained in the following way. Their progressive hepatic failure probably impairs the normal hepatic destruction of glucocorticoids, whereby the plasma [cortisol] is rising. Such patients should be controlled, because the liver destruction is likely to proceed, and they become more and more Cushingoid with continued alcohol abuse. 7. Congenital adrenal hyperplasia This is an autosomal, recessive enzyme deficiency blocking steroid synthetic pathway of the adrenal cortex. The most frequent of these rare genetic disorders is the 21-hydroxylase deficiency on chromosome 6. Lack of hydroxylase blocks the conversion of 17-hydroxyprogesterone into 11- deoxycortisol and further to cortisol. Instead androstenedione and testosterone is produced and virilization is an inevitable result. The impaired cortisol release from the adrenal increases the pituitary ACTH secretion by negative feedback and more precursor molecules are accumulated (ie, hydroxyprogesterone, androstenedione and testosterone). The clinical features are those of an adrenogenital syndrome. Newborn girls may show clitoral hyperplasia and labioscrotal fusion, and later some boys develop precocious puberty. Varying degrees of virilism in females, birdied ladies in circus, or perhaps only hirsutism before puberty occur. The lack of aldosterone causes salt-and water-loss, which may be life threatening. A defect in the 11-hydroxylase enzyme gene also leads to overproduction of androgens and 11-deoxycorticosterone from accumulated precursors. The later has a massive mineralocorticoid effect. The therapy is exact replacement of glucocorticoid activity and any other inefficiency. Primary hypoadrenalism is a complete destruction of the adrenal cortex, and thus it impairs all three lines of steroid production (ie, glucocorticoids, mineralocorticoids and sex hormones). Primary hypoadrenalism occurs in an acute and a chronic form. Acute hypoadrenalism (ie, adrenal crisis or Waterhouse-Friderichsens syndrome) is a fulminant infection with shock and massive bleeding all over. The large internal bleedings are visible at the skin as areas of purpura. The patient dies within a few hours, if not treated urgently. Hydrocortisone (100 mg) is given intramuscularly. Antibiotics and cortisol is administered. Chronic hypoadrenalism (ie, Addison’s disease) is hypocorticism due to destruction with atrophy of the entire adrenal cortex by autoimmune processes, malignancy, infarction or infection. Most cases develop organ-specific autoantibodies; these cases are associated with many other autoimmune disorders (eg, diabetes mellitus, hypoparathyroidism, pernicious anaemia, vitiligo and thyroiditis). Addison’s disease is a life-threatening condition with loss of Na+ and thus also of ECV. Symptoms and signs include: reduced blood pressure, reduced blood [glucose], tiredness and skin pigmentation caused by MSH, whose level is increased concurrent with the overproduction of ACTH, due to the decreased negative feedback. Severe hypotension may develop into cardiovascular collapse, which is called an Addisonian crisis. The clinical manifestations are consequences of the impaired secretion of the three hormone groups (Fig. 30-10).
Fig. 30-10: Clinical manifestations of primary, chronic adrenocortical insufficiency (Addison). The low cortisol increases the hypothalamic CRH secretion through feedback and thereby increases ACTH secretion. Fragments of the ACTH molecule form MSH, which is responsible for the skin hyperpigmentation. In the absence of glucocorticoids renal phsophate excretion is depressed. When Addison’s disease is suspected, a single plasma ACTH level is often diagnostic, as it will be elevated if hypocorticism originates in the adrenal gland (primary) and normal or low in hypothalamic-pituitary hypocorticism (secondary). - Differentiation between the hypothalamic and the pituitary hypocorticism can be accomplished by injection of CRH, which stimulates the pituitary directly. - Insulin-induced hypoglycaemia normally recruits the combined hypothalamic-pituitary axis. Hypoglycaemia is a prolonged stress, which will normally increase ACTH and glucocorticoid secretion. If there is adrenal gland insufficiency then this increase in glucocorticoid secretion is not observed. Hypothalamic and pituitary failure is detected when a dose of CRH restores glucocorticoid release. - A single ACTH-injection along with measurement of plasma [cortisol] is a simple screening procedure for adequate endogenous ACTH secretion. Synthetic ACTH is injected intravenously. If plasma [cortisol] is normal 30 minutes later, the disease is not a primary adrenocortical atrophia. - The metyrapone test is used when patients are suspected to have Addison’s hypocorticism, but have reacted normally to the ACTH-test. Metyrapone inhibits the last step in cortisol synthesis, so 11-dehydrocortisol is accumulated and an acute cortisol deficiency is created. Plasma-[Cortisol] and -[11-deoxycortisol] is measured two mornings in succession, and the adrenal 11-hydroxylase is inhibited with 500 mg metyrapone every second hour during 24 hours. The plasma- [cortisol] must fall while -[11-deoxycortisol] accumulates. The fall in cortisol biosynthesis increases the CRH and ACTH secretion by negative feedback, provided the hypothalamic-pituitary system is intact. Failure to respond to metyrapone suggests a hypothalamic-pituitary defect that has abolished this feedback. Substitution therapy with gluco- and mineralo-corticoids is necessary for the rest of the patient’s life. This is usually caused by prolonged corticoid therapy of chronic disorders such as rheumatoid arthritis or COLD. A rare form of secondary hypoadrenalism is panhypopituitarism. Rarely some patients have attacks of severe hypertension., which are caused by a medullary tumour (a phaeochromocytoma of chromaffine cells), which liberates large amounts of catecholamines. I. Each of the following five statements have False/True options: A. Atropine blocks muscarinic receptors, and d-tubocurarine blocks nicotinic receptors. B. b-Receptors are blocked by propranolol. C. Catecholamines have a half-life in plasma of more than 20 min. D. The three important catecholamines in humans are adrenaline, noradrenaline, and DOPA. E. b1-receptors are stimulated by adrenaline and located in the myocardium. II. Each of the following five answers have False/True options: ACTH stimulates the liberation of: A. Hydrocortisone or cortisol B. Adrenaline C. Aldosterone D. Adrenal androgens E. Dopamine. A man, 49 years of age (height 1.86 m; weight 62 kg) is in hospital due to the following symptoms and signs. He is nervous and has a diffuse struma. A characteristic, blowing sound is heard from the thyroid gland with a stethoscope. The blood pressure is 145/70 mm Hg. An attack of cardiac arrhythmia is recorded with an ECG. A P-wave frequency above 400 per min is present during the attack. The concentration of thyroxine in blood serum is 180 nM. The distribution volume for radioactive thyroxine is 8 l. The elimination rate of this thyroxine is 14% of the total content per 24 hours. 1. Calculate the serum [thyroxine] in mg per l. 2. How much thyroxine is eliminated per 24 hours expressed in mg daily? 3. Calculate the thyroid plasma clearance of iodide, when the concentration of free iodide in plasma is 4 mg per l. 4. Explain the condition of this patient. A 22-year-old medical student is treated with an intravenous dose of PTH. This changes his renal excretion flux of two substances and their plasma concentrations. 1. Describe the alterations and explain the mechanisms. 2. What is the diagnosis? 3. Describe the most likely symptoms and signs of this patient before treatment. A female (52 years of age; height 1.68 m; weight 62 kg) is in hospital due to her third attack of kidney stone pains. The first routine examination with arterial blood analysis reveals the following. Her blood pH is 7.21 and her plasma [Ca2+] is 2 mmol per l (mM) in ionised form. Her ionised [Ca2+] constitute 62% of the total calcium concentration in plasma. The inorganic phosphate concentration (total) is 0.84 mmol per l of plasma. The patient excretes 2-3 l of urine per day. pK2 = 6.8 for H3PO4. 1. Calculate the fractions of primary (H2PO4-) and secondary (HPO4--) phosphate in her plasma. Compare the results to the normal mean value of 1 mM of inorganic phosphate with 20% primary and 80% secondary phosphate at pH = 7.40. 2. Calculate the total plasma [calcium] of this patient and compare the result to the normal mean value of 2.5 mM. 3. Why is the ionised calcium fraction much higher than normal (0.45)? 4. Could this condition be the result of a classical endocrine disease? 5. Why did this patient develop kidney stones? Was her diuresis normal? If not explain why. Try to solve the problems before looking up the answers. · Growth hormone (GH) produced in the placenta differs from the pituitary GH by a few amino acid residues. Placental GH suppresses release of maternal, pituitary GH during pregnancy. Placental GH stimulates maternal metabolism and foetal cell proliferation and hypertrophia. · Foetal thyroid hormones stimulate brain development, and foetal insulin stimulates foetal growth, cellular glucose uptake and glucose utilisation. Paracrine and autocrine growth factors are also important for foetal growth: Insulin-like growth factor-II (IGF-II), nerve growth factors (NGF), epidermal growth factor (EGF), and platelet derived growth factor (PDGF). · Growth hormone and insulin are the most important anabolic hormones in the human body. · PTH binds to membrane receptors on target cells in bone, kidney and gut. The PTH actions result in hypercalcaemia and hypophosphataemia. The bone resorption is illustrated by a high basic phosphatase concentration in blood. · Cortisol stimulates hepatic glucose production, both glycogenolysis and gluconeogenesis, lipolysis, formation of FFA and of ketone bodies. Cortisol inhibits the glucose uptake in target cells (GLUT 4 in muscle cells, heart cells and adipocytes). · Therapeutic doses of glucocorticoids are used for a multitude of diseases such as inflammations, allergy, malignancy and aplastic anaemia. The negative effects are delayed healing of wounds and increased gluconeogenesis with destruction of tissue proteins. · Aldosterone is the major mineralocorticoid with corticosterone contributing only little. Aldosterone promotes the reabsorption of Na+ and increases the secretion of K+ and H+ in the distal tubular system (ie the cortical collecting ducts and the connecting segment). A rise in serum - [K+] from normal or a fall in serum- [Na+] releases aldosterone. The renin-angiotensin-aldosterone cascade controls the adrenal aldosterone secretion, not ACTH. · Sex steroids are mainly weak adrenal androgens, which are metabolised to testosterone and dihydrotestosterone. Also a small oestrogen production is present in healthy persons. · Catecholamines increase the heart rate and the cardiac output by stimulation of the adrenergic b1-receptors in the myocardium. · Noradrenergic nerve fibres innervate vessels all over the body, and this system usually has some tonic, vasoconstrictor activity. The a1-receptors are located on the surface of vascular smooth muscles. · Catecholamines dilatate the bronchial airways by stimulation of their adrenergic b2-receptors. Catecholamines increase both tidal volume and respiratory frequency. The result is an increase in ventilation together with an increase in cardiac output. · Catecholamines relax the smooth muscles of the digestive tract (b2-receptors), but contract the sphincters just like the sympathetic nerve system. · Catecholamines stimulate metabolism (T3). Adrenaline stimulates hepatic glycogenolysis and lipolysis in adipose tissue. Adrenaline increases the plasma concentrations of glucose, FFA and ketoacids. · Adrenaline stimulates the ascending reticular system (ie, the reticular activating system or RAS) in the brain stem, keeping us alert and causing arousal reactions with desynchronisation of the EEG. · Pituitary acidophilic adenomas that secrete excess growth hormone (GH) causes gigantism in children and acromegaly in adults. Rare cases are caused by GHRH excess release from the hypothalamus. Since pituitary acidophilic adenomas often contain both somatotropic and mammotropic cells, the combined adenomas secrete an excess of both GH and prolactin (causing galactorrhoea in males). · Primary parathyroid hyperfunction is almost inevitably due to parathyroid adenomas or hyperplasia that secrete an excess of parathyroid hormone, PTH. Ectopic tumours have been found in the mediastinum and elsewhere. Excessive secretion of PTH leads to: bone resorption, high [Ca2+] in plasma, high Ca2+ -excretion in the kidneys with renal stone formation, bone lesions, and metastatic calcification. · Chronic hypoadrenalism (ie, Addison’s disease) is hypocorticism due to destruction with atrophy of the entire adrenal cortex by autoimmune processes, malignancy, infarction or infection. Most cases develop organ-specific autoantibodies; these cases are associated with many other autoimmune disorders (eg diabetes mellitus, hypoparathyroidism, pernicious anaemia, vitiligo and thyroiditis). Costanzo, L S. Regulation of calcium and phosphate homeostasis. Am. J. Physiol. 275 (Adv. Physiol. Educ.): S206-S216, 1998. Änggård, E. "Nitric oxide: mediator, murderer, and medicine." Lancet 343: 1199-1206, 1994. Tonegawa, S. "The molecules of the immune system," Sci. Am. 253: 122-131, 1985.
|
||









.jpg){kind=link}
{kind=link}
Click here to introduce your comments or contributions